Newborn Screening
Neonatal screening represents one of the most successful public health initiatives of the 20th century. Thanks to newborn screening, approximately 15,000 newborns are identified in the US each year for which screening, diagnosis, and effective treatments can be used early in life to significantly impact the child’s quality of life and outcome. What began as a targeted effort to prevent severe intellectual disability in a single, rare genetic disorder eventually paved the way for the comprehensive newborn screening panels used worldwide today.
The First Screen
The original use case for neonatal screening was the early detection of Phenylketonuria (PKU). PKU is a rare, inherited metabolic disorder in which the body lacks the enzyme needed to break down the amino acid phenylalanine. Before screening existed, infants with PKU appeared healthy at birth. However, as they consumed breast milk or formula (both of which contain phenylalanine), the amino acid would build up to toxic levels in their blood, leading to severe, irreversible brain damage and intellectual disability.
Eventually, it was discovered that if an infant with PKU was placed on a strict, low-phenylalanine diet immediately after birth, they would develop normally. The challenge, however, was diagnosing the condition before the neurological damage occurred. Early urine tests (using ferric chloride) were unreliable in the first weeks of life, often missing the crucial window for intervention.
Around 1961, Dr. Robert Guthrie, a medical microbiologist conducting cancer research at the Roswell Park Cancer Institute and the father of a child with intellectual disabilities, developed a simple, inexpensive “bacterial inhibition assay.” He discovered that he could test for elevated phenylalanine levels using a few drops of capillary blood collected from a newborn’s heel prick. This blood was dried on special filter paper—now universally known as the “Guthrie card”—making it easy to transport samples from maternity wards to centralized laboratories.
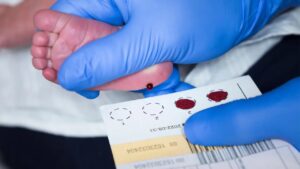
The concept of newborn screening for PKU was resisted by the New York public health labs, believing that the incidence of PKU was too low to warrant the effort, but Dr. Guthrie found an ally in Dr. Robert MacCready, Director of the Diagnostic Division of the Massachusetts Public Health Laboratory. Following successful large-scale trials, Massachusetts became the first US state to establish universal newborn screening for PKU in the Fall of 1962. Soon, several other states, including Oregon, Ohio, and Maryland, as well as the New York Regional Public Health Laboratory in Buffalo, began PKU screening. But the road to universal screening was bumpy, there was opposition from physicians and medical societies that considered it to be “socialized medicine” and an infringement on the private practice of medicine. Howver, by the 1970s, almost every state had implemented mandatory PKU screening, and the practice rapidly spread internationally.
One Test, One Disease
The “one test, one disease” era, which stretched from Dr. Robert Guthrie’s breakthrough in the early 1960s until the widespread adoption of tandem mass spectrometry (MS/MS) in the late 1990s, was defined by a strict logistical ceiling. During this period, every time a health department wanted to add a new condition to the newborn screening panel, laboratories had to adopt an entirely new, distinct analytical method. This required a separate punch from the dried blood spot, different chemical reagents, and dedicated laboratory equipment and technicians. Because of this physical and financial bottleneck, the expansion of newborn screening was slow and methodical, but many states added screening for some combination of the following.
- Galactosemia: a disorder where infants cannot metabolize galactose (a sugar found in breast milk and cow’s milk formula), leading to liver failure, cataracts, and brain damage. 1970s.
- Maple Syrup Urine Disease (MSUD) & Homocystinuria: amino acid disorders that cause severe disabilities if untreated. 1970s.
- Congenital Hypothyroidism (CH): leads to severe physical and mental disabilities if undetected. Mid-1970s.
- Hemoglobinopathies, primarily Sickle Cell Disease (SCD): testing for hemoglobinopathies required a technology known as electrophoresis. 1970s.
- Congenital Adrenal Hyperplasia (CAH): an endocrine disorder that can cause fatal salt-wasting crises in newborns. 1980s.
- Biootinidase Deficiency: an enzyme defect which can lead to seizures and hearing loss if undetected. 1980s.
By the early 1990s, a well-funded state laboratory might be screening for roughly 5 to 8 conditions. To do so, a technician had to take the Guthrie card and punch out 5 to 8 separate tiny circles of dried blood, distributing them to entirely different departments in the lab—one running bacteria culture plates, one running electrophoresis gels, and another running immunoassays. The system was at its maximum physical capacity, setting the stage for the application of a new technology in the 1990s that was more scaleable (tandem mass spectrometry).
Tandem Mass Spectrometry (MS/MS)
Instead of using a biological reaction to find a single problem, MS/MS identifies compounds based on their specific mass and charge. The machine separates ions, fragments them, and analyzes the pieces to identify the specific “fingerprint” of various molecules.

The key advantage of MS/MS for newborn screening was multiplexing. A single punch of a dried blood spot could now be analyzed to detect dozens of different metabolites (amino acids and acylcarnitines) simultaneously. After an initial investment in mass spectrometry equipment, state screening programs could jump from checking for around 5 conditions to over 30 conditions almost overnight, without changing the established hospital processes for collecting and shipping newborn heelstick blood spots on “Guthrie cards” to state labs.
In 2006, the American College of Medical Genetics (ACMG) recommended a uniform panel of 29 core conditions for the U.S., reducing the patchwork of state-by-state variation. The Recommended Uniform Screening Panel (RUSP) was legislatively mandated in 2008 and implemented in 2010 by the Secretary of HHS and became the policy benchmark.
More recently, next-generation sequencing has been incorporated into newborn screening, enabling detection of a far wider range of genetic conditions. Programs like “BabySeq” have explored genome-wide sequencing of newborns. Genetic conditions like cystic fibrosis (CF), spinal muscular atrophy (SMA),, and severe combined immunodeficiency (SCID) have been added to the RUSP, where early treatment is crucial. The U.S. RUSP now includes 40 core conditions and 26 secondary disorders, as well as screening for congenital heart disease and hearing loss.
Newborn screening (NBS) programs are implemented and managed at the state or territorial level. While all 56 NBS programs are tracking the RUSP, changes to the RUSP occur frequently, not all NBS programs adopt every change, adoption of some changes takes longer for some programs than for others, and some programs add tests that are not part of the RUSP. There is no central or integrated system for laboratory testing of newborn blood spots, so the 56 NBS programs use the services of an array of NBS public health laboratories and privately run laboratories.
Although it is now technically and financially feasible, the prospect of universal genetic sequencing continues of newborns to be debated among physicians, ethicists, and scientists:
- Sequencing often uncovers genetic variants of unknown significance (and perhaps no significance)
- The ethical tension between early knowledge and parental anxiety for conditions with no immediate treatment
- Comprehensive genetic screening remains unavailable in much of the world and likely to remain so for many years to come
The Evolution of Newborn Screening
| Era | Primary Technology | Key Limitation | Typical Panel Size |
| 1960s-1980s | Bacterial Inhibition | One test per disease | 1-3 conditions |
| 1980s-early 1990s | Immunoassays and Electrophoresis | Labor-intensive, slow expansion | 5-8 conditions |
| late 1990s’-present | Tandem Mass Spectrometry | High initial equipment cost | 30-60+ conditions |
| 2010s-present | Next-generation sequencers | High initial equipment cost | Selected disorders, some state-by-state variation |
Recommended Universal Screening Panel (RUSP)
| Disorder | Incidence per 100,000 Live Births | Category | Notes |
|---|---|---|---|
| Organic Acid Conditions | |||
| 3-Hydroxy-3-methylglutaric aciduria (HMG) | < 1 | OA | |
| 3-Methylcrotonyl-CoA carboxylase deficiency (3-MCC) | 2–4 | OA | |
| Beta-ketothiolase deficiency (BKT) | < 1 | OA | |
| Glutaric acidemia type I (GA1) | 2–4 | OA | |
| Holocarboxylase synthase deficiency (HCS) | < 1 | OA | |
| Isovaleric acidemia (IVA) | 1–2 | OA | |
| Methylmalonic acidemia – cobalamin disorders (Cbl A,B) | 1–2 | OA | |
| Methylmalonic acidemia – methylmalonyl-CoA mutase (MUT) | 2–3 | OA | |
| Propionic acidemia (PA) | 2–3 | OA | |
| Fatty Acid Oxidation Conditions | |||
| Carnitine uptake defect / primary carnitine deficiency (CUD) | 1–3 | FAO | |
| Long-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency (LCHAD) | 1–2 | FAO | |
| Medium-chain acyl-CoA dehydrogenase deficiency (MCAD) | 10–20 | FAO | Most common FAO disorder |
| Trifunctional protein deficiency (TFP) | < 1 | FAO | |
| Very long-chain acyl-CoA dehydrogenase deficiency (VLCAD) | 1–3 | FAO | |
| Amino Acid Disorders | |||
| Argininosuccinic aciduria (ASA) | 1–2 | AA | |
| Citrullinemia type I (CIT) | 1–2 | AA | |
| Classic phenylketonuria (PKU) | 8–10 | AA | Original Guthrie test target |
| Homocystinuria (HCY) | < 1 | AA | |
| Maple syrup urine disease (MSUD) | 2–4 | AA | |
| Tyrosinemia type I (TYR I) | 1–2 | AA | |
| Endocrine Disorders | |||
| Congenital adrenal hyperplasia (CAH) | 10–15 | EN | |
| Primary congenital hypothyroidism (CH) | 25–40 | EN | Most common endocrine disorder screened |
| Hemoglobin Disorders | |||
| S,S disease – sickle cell anemia | ~40–50 (combined SCD) | HB | Higher prevalence in African-ancestry populations |
| S,C disease (HbSC) | ~40–50 (combined SCD) | HB | Included in combined SCD estimate |
| S,beta-thalassemia (HbS/BT) | ~40–50 (combined SCD) | HB | Included in combined SCD estimate |
| Other Core Conditions | |||
| Biotinidase deficiency (BIOT) | 5–7 | OT | |
| Classic galactosemia (GALT) | 4–8 | OT | |
| Critical congenital heart disease (CCHD) | 100–200 | OT | Screened via pulse oximetry, not blood spot |
| Cystic fibrosis (CF) | 25–35 | OT | |
| Glycogen storage disease type II – Pompe disease (GSD II) | 2–4 | OT | |
| Guanidinoacetate methyltransferase deficiency (GAMT) | < 1 | OT | |
| Hearing loss (HL) | 150–200 | OT | Screened via audiologic testing |
| Infantile Krabbe disease | 1–2 | OT | |
| Mucopolysaccharidosis type I – Hurler syndrome (MPS I) | 1–4 | OT | |
| Mucopolysaccharidosis type II – Hunter syndrome (MPS II) | 1–3 | OT | |
| Severe combined immunodeficiency (SCID) | 1–3 | OT | |
| Spinal muscular atrophy (SMA) | 1–2 | OT | |
| X-linked adrenoleukodystrophy (X-ALD) | 1–4 | OT | |
| Duchenne muscular dystrophy (DMD) * | 20–30 | OT | * Added December 2025 |
| Metachromatic leukodystrophy (MLD) * | 1–2 | OT | * Added December 2025 |
| Total Core Conditions: 40 |
|||
Notes:
Most common conditions (incidence ≥ 25/100,000): hearing loss (~150–200), critical congenital heart disease (~100–200), congenital hypothyroidism (~25–40), cystic fibrosis (~25–35), and Duchenne muscular dystrophy (~20–30).
Moderately common (5–25/100,000): MCAD deficiency, PKU, congenital adrenal hyperplasia, sickle cell disease subtypes, and biotinidase deficiency.
Rare (< 5/100,000): most of the organic acid conditions, lysosomal storage diseases (Pompe, MPS I, MPS II, Krabbe), SCID, SMA, X-ALD, and GAMT deficiency.
Incidences shown above are for the general population, and may vary in specific populations — sickle cell disease, for example, is far more common in infants of African ancestry, while PKU varies by ethnicity. Sources are the CDC MMWR (2020), APHL NewSTEPs data (2023), and HRSA.
References
- Guthrie, R., & Susi, A. (1963). A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics, 32(3), 338-343.
- Levy, H. L. (2021). Robert Guthrie and the Trials and Tribulations of Newborn Screening. International Journal of Neonatal Screening, 7(1), 5. https://doi.org/10.3390/ijns7010005
- Watson MS, Lloyd-Puryear MA, Howell RR (2022): The Progress and Future of US Newborn Screening. International Journal of Neonatal Screening, 8, 41. https://doi.org/10.3390/ijns8030041
- Watson MS, Mann MY, Lloy-Puryear MA, Rinaldo P, Howell RR (2006): Newborn Screening: Toward a Uniform Screening Panel and System. Genet Med 2006 May; 8 Suppl 1: 1S-252S doi: 10.1097/01.gim.0000223891.82390.ad. https://pubmed.ncbi.nlm.nih.gov/16783161/
- Therrell BL, Padilla CD, Borrajo GJC et al (2024): Current Status of Newborn Bloodspot Screening Worldwide 2024: A Comprehensive Review of Recent Activities. Int J Neonatal Screen. 2024 May 23;; 10(2):38. doi: 10.3390/ijns10020038. https://pmc.ncbi.nlm.nih.gov/articles/PMC11203842/
- Singh S, Ojodu J, Kemper AR, Lam WKK, Grosse SD (2023). Implementation of Newborn Screening for Conditions in the United States First Recommended during 2010-2018. Int J Neonatal Screen. 2023 Apr 6;9(2):20. doi: 10.3390/ijns9020020. PMID: 37092514; PMCID: PMC10123615. https://pmc.ncbi.nlm.nih.gov/articles/PMC10123615/
- National Academies of Sciences, Engineering, and Medicine. (2022). Newborn Screening in the United States: A Vision for Sustaining and Expanding a Vital Public Health System. Washington, DC. https://www.nationalacademies.org/projects/HMD-HSP-23-14/publication/29102 (free download)
- Health Resources and Services Administration (HRSA) — Recommended Uniform Screening Panel: hrsa.gov/advisory-committees/heritable-disorders/rusp
- National Organization for Rare Disorders (NORD) Newborn Screening Policy Page (2025): rarediseases.org/policy-issues/newborn-screening
- NewSTEPs State-by-State Data Dashboard: newsteps.org/resources/data-visualizations
- EveryLife Foundation — RUSP Alignment Legislation by State: everylifefoundation.org
Last Updated on 03/05/26